hicrep# #的介绍
Hi-C数据分析和解释仍处于早期阶段。特别是,一直缺乏可靠的统计指标来评估Hi-C数据的质量。当无法获得生物重复时,研究人员通常依赖于Hi-C相互作用热图的视觉检查或检查远程相互作用读对与总测序读对的比例,这两种方法都没有可靠的统计数据支持。当有两个或两个以上的生物重复时,通常的做法是计算两个Hi-C数据矩阵之间的Pearson或Spearman相关系数,并将其用作质量控制的度量。然而,这种过于简化的方法是有问题的,可能会导致错误的结论,因为它们没有考虑Hi-C数据的独特特征,如距离依赖和域结构。因此,两个不相关的生物样本可以具有很强的Pearson相关系数,而两个视觉相似的重复可以具有较差的Spearman相关系数。不相关样本之间的Pearson和Spearman相关性高于真实生物重复之间的Pearson和Spearman相关性也并不罕见。
我们开发了一个新的框架,hicrep,用以评估Hi-C数据的再现性。该方法首先通过平滑Hi-C矩阵来最小化噪声和偏差的影响,然后根据Hi-C数据的基因组距离对其进行分层来解决距离依赖效应。我们进一步采用层调相关系数(SCC)作为Hi-C数据再现性的度量。SCC的取值范围为-1 ~ 1,可用于比较再现性的差异程度。我们的框架还可以推断SCC的置信区间,并进一步估计不同数据集的可重复性测量差异的统计显著性。
在这个小插图中,我们解释了方法的基本原理,并提供了使用的指导hicrep以评估Hi-C染色体内复制的可重复性。
请引用以下论文:
HiCRep:使用层调整相关系数评估Hi-C数据的可重复性。杨涛,张飞鹏,Galip Gurkan Yardimci, Ross C Hardison, William Stafford Noble,岳峰,李群华。bioRxiv 101386;doi:https://doi.org/10.1101/101386
下载源包hicrep_0.99.5.tar.gz从Github。或者从Bioconductor安装。
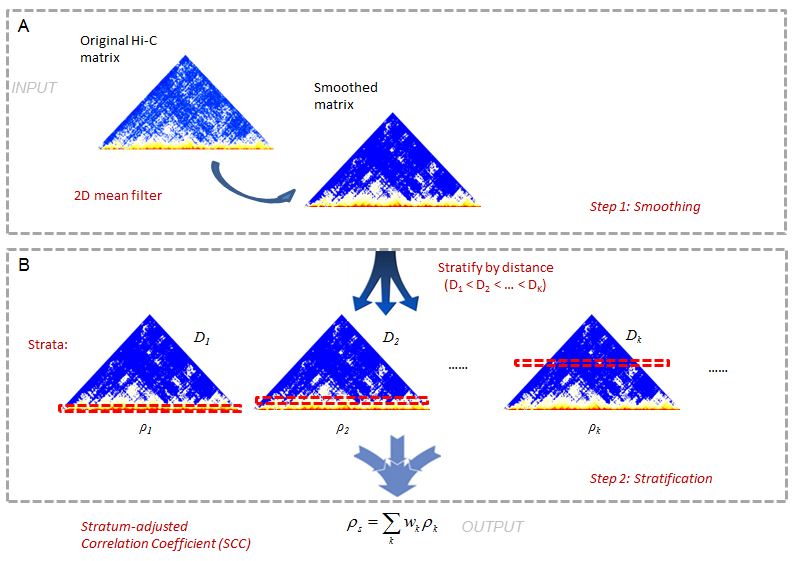
这是一个两步方法(图1)。在Hi-C数据中,通常很难实现足够的覆盖。当样本序列不够时,欠采样引入的局部变化会使捕获大域结构变得困难。为了减少局部变异,在评估再现性之前,我们首先平滑接触图。虽然平滑滤波器会降低单个空间分辨率,但它可以提高相互作用增强的区域的连续性,从而增强域结构。我们使用二维移动窗口平均滤波器平滑Hi-C接触图。这种选择是为了平均滤波器计算简单、快速,以及Hi-C隔室的矩形形状。
在第二步中,我们将Hi-C读数按接触位点的距离进行分层,计算各层间的Pearson相关系数,然后将各层相关系数汇总成一个聚合统计量。我们称之为地层调整相关系数(SCC)。具体方法请参考我们的手稿。
(图1。hicrep管道示意图)(https://raw.githubusercontent.com/MonkeyLB/hicrep/master/vignettes/hicrep-pipeline.JPG)
输入仅仅是两个要比较的Hi-C矩阵。Hi-C矩阵应该有维数\ (N (3 + N) \ \倍).附加的三个初始列是染色体名称和两个接触箱的中点坐标。在这里,我们应该有一个示例数据:
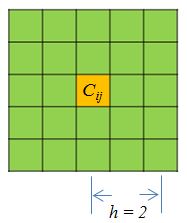
## [1] 52 55# # V1 V2 V3 V4 V5 V6 V7 V8 V9 V10 # # 1 chr22 0 1000000 0 0 0 0 0 0 0 # # 2 chr22 1000000 2000000 0 0 0 0 0 0 0 # # 3 chr22 2000000 3000000 0 0 0 0 0 0 0 # # 4 chr22 3000000 4000000 0 0 0 0 0 0 0 # # 5 chr22 4000000 5000000 0 0 0 0 0 0 0 # # 6 chr22 5000000 6000000 0 0 0 0 0 0 0 # # 7 chr22 6000000 7000000 0 0 0 0 0 0 0 # # 8 chr22 7000000 8000000 0 0 0 0 0 0 0 # # 9 chr22 8000000 9000000 0 0 0 0 0 0 0 # # 10 chr22 9000000 10000000 0 0 0 0 0 0 0单一函数准备对给定邻域大小参数的HiC矩阵进行变换,平滑数据\ \ (h),并过滤两个重复中计数为零的箱子。参数包括两个矩阵,矩阵的分辨率,平滑参数,以及考虑的最大距离。分辨率就是箱子的大小。平滑参数决定了平滑的邻域大小。下图(图2)演示了一个点的平滑邻域\ (C_ {ij} \):
[Figure2。一个平滑的社区\(h = 2\)) (https://raw.githubusercontent.com/MonkeyLB/hicrep/master/vignettes/Smoothing_neighborhood.JPG)
最大距离是超过这个距离的接触者将被调查。这里我们展示了一个分辨率为40kb的例子,平滑参数\(h = 1\),以及距离超过5M的接触将不被考虑:
## v1 v2 v3 v4 ## 796 15500000 15500000 314.666667 515.11111 ## 797 15500000 16500000 539.88889 871.11111 ## 798 15500000 17500000 579.66667 927.11111 ## 799 15500000 18500000 283.00000 439.44444 ## 800 15500000 19500000 65.33333 95.55556 ## 801 15500000 20500000 32.88889 53.88889输出的前两列是两个接触箱的中点坐标,最后两列是平滑的(如果没有)\(h = 0\))的值。
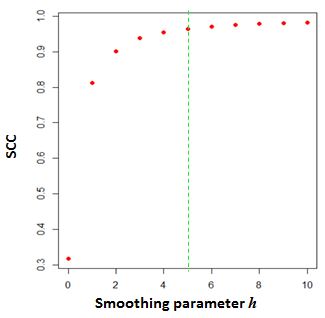
一个自然的问题是如何选择平滑参数\ \ (h)为了选择\ \ (h)客观上,我们开发了一种搜索最优平滑参数的启发式程序。我们的程序是基于这样的观察设计的,即复制样本的接触图之间的相关性首先随着平滑程度的增加而增加,当达到足够的平滑时,就会达到平稳期。接下来,我们使用一对合理的深度序列交互图作为训练数据。我们将数据随机划分为10%的10个分数,然后计算采样数据的SCC,每个分数在一系列平滑参数上按升序排列,然后选择平均再现性得分增量小于0.01的最小h。这个过程重复十次,从这十个h中选择一个模式。
# #平滑:0# #平滑:1# #平滑:2## htrain中的警告(HiCR1, HiCR2, 1e+06, 5e+06, 0:2):注意:可能是您的搜索范围太窄。##尝试扩大范围并重新运行它## [1](图3)。选择最优的平滑参数\ \ (h)) (https://raw.githubusercontent.com/MonkeyLB/hicrep/master/vignettes/Smoothing_parameter.JPG)
上图显示了SCC随时间的变化\ \ (h)从0增加到10。的参数\ (h = 5 \)作为最优平滑邻域大小。
平滑参数的选择可能会受到排序深度的影响。序列深度数据不足可能导致平滑邻域大小膨胀。我们建议使用足够的Hi-C序列数据在给定分辨率(即>整个染色体的总读数为3亿)的情况下训练平滑参数。为了比较具有相同分辨率的复制对之间的SCC,应该使用相同的平滑参数。
在前一节中,我们提到测序深度可能是一种混杂效应。如果两个重复的reads总数相差很大,建议对测序深度较高的进行下采样,使其与测序深度较低的相同。最好的方法是使用bam文件进行子抽样。如果你只有矩阵文件可用,我们做了一个函数来从矩阵文件中进行向下采样。
## [1] 2065436## [1] 2e+05当out = 0时,函数将返回原始格式的矩阵。当它等于1时,将输出向量化格式。
在对原始矩阵进行预处理之后,我们就可以计算SCC及其方差了。输入文件只是函数的输出数据集(即,向量化、平滑化、过滤零)准备.我们还需要指定矩阵的分辨率,以及考虑的最大接触距离。
## [,1] ## [1,] 0.9822897## [1] 0.004437169## R版本3.6.1(2019-07-05)##平台:x86_64-pc-linux-gnu(64位)##运行在Ubuntu 18.04.3 LTS ## ##矩阵产品:默认## BLAS: /home/biocbuild/bbs-3.10-bioc/R/lib/libRblas。所以## LAPACK: /home/biocbuild/bbs-3.10-bioc/R/lib/libRlapack。所以## ## locale: ## [1] LC_CTYPE=en_US。UTF-8 LC_NUMERIC= c# # [3] LC_TIME=en_US。UTF-8 LC_COLLATE= c# # [5] LC_MONETARY=en_US。utf - 8 LC_MESSAGES = en_US。UTF-8 ## [7] LC_PAPER=en_US。UTF-8 LC_NAME= c# # [9] LC_ADDRESS=C lc_phone = c# # [11] LC_MEASUREMENT=en_US。UTF-8 LC_IDENTIFICATION=C ## ##附加的基本包:## [1]stats graphics grDevices utils datasets methods base ## ##其他附加包:## [1]hicrep_1.10.0 ## ##通过命名空间加载(且未附加):## [1]compiler_3.6.1 magrittr_1. 1.5 htmltools_0.4.0 tools_3.6.1 ## [5] yaml_2.2.0 Rcpp_1.0.2 stringi_1.4.3 rmarkdown_1.16 ## [9] knitr_1.25 string_1 .4.0 digest_0.6.22 xfun_0.10 ## [13] rlang_0.4.1 evaluate_0.14输出结果列表,包括特定地层的Pearson相关性、权重系数、SCC和SCC的渐近标准差。最后两个数字是我们在大多数情况下需要的。
计算速度由染色体的大小决定。对于22号染色体的数据,htrain大概需要15分钟,还有get.scc大约需要1~2分钟。对于1号染色体,get.scc大约需要90分钟。该估算基于Intel i7-3250M内核。
{kind=link}
{kind=link}
{kind=link}